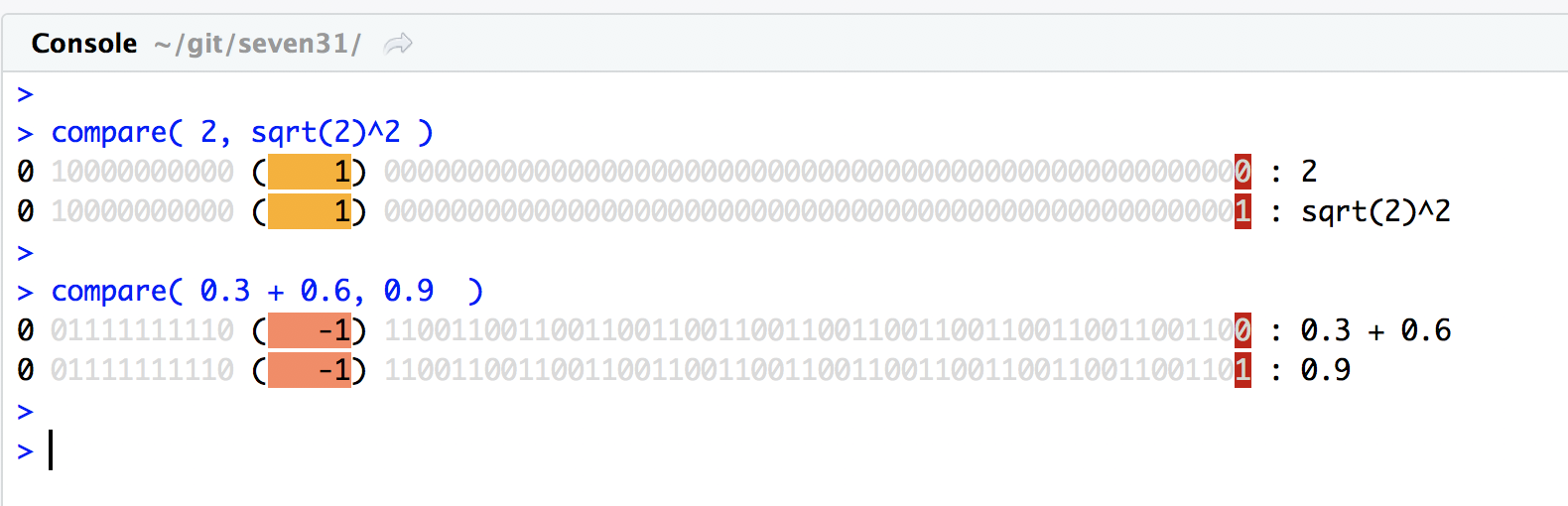
Preparing data is the most time consuming part of of data analysis
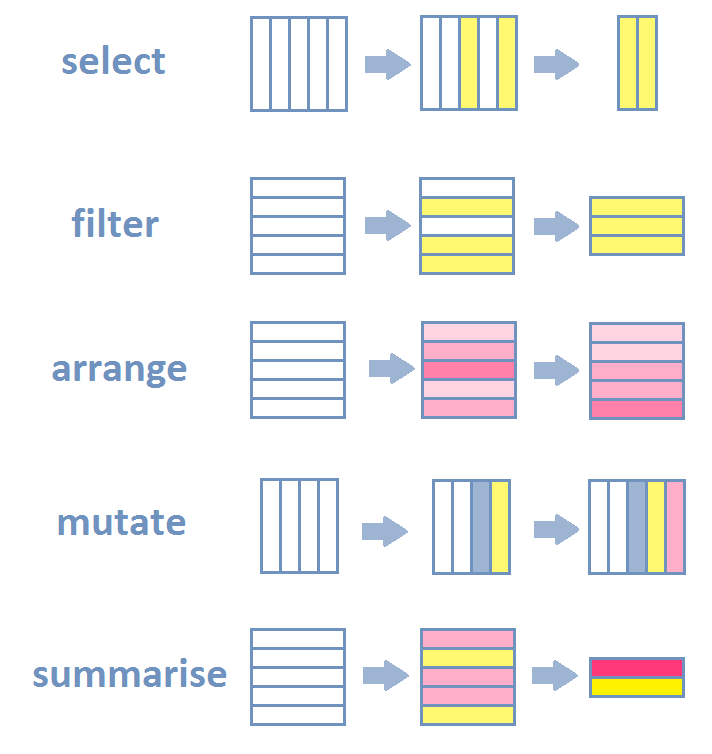
dplyr is a tool box for working with data in tibbles/data frames
- The most import data manipulation operations are covered
- Selection and manipulation
- Selection and manipulation of
- observations,
- variables and
- values.
- Summarizing
- Grouping
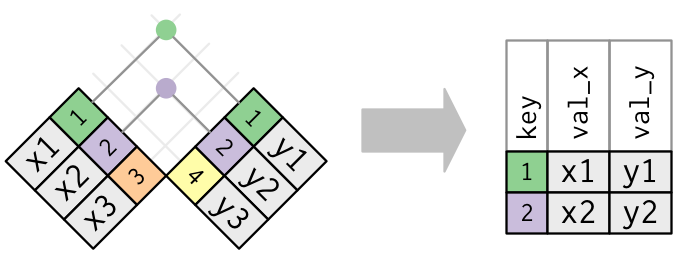
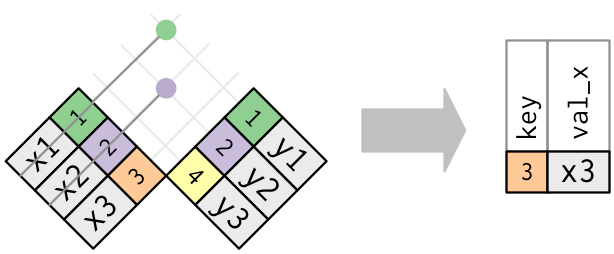
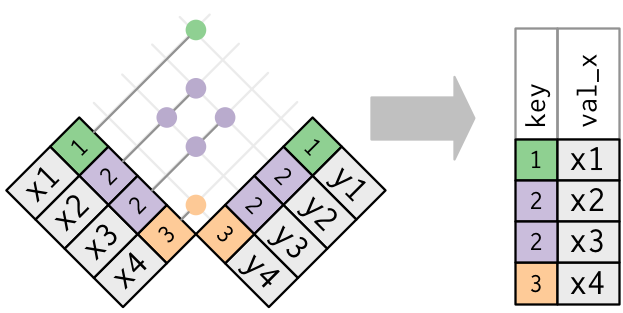
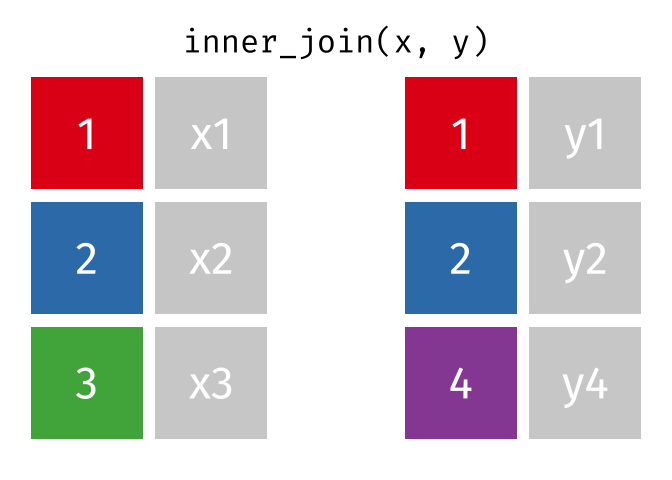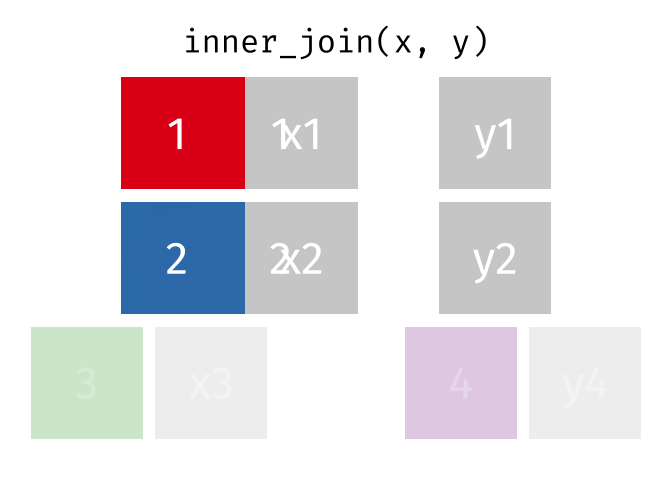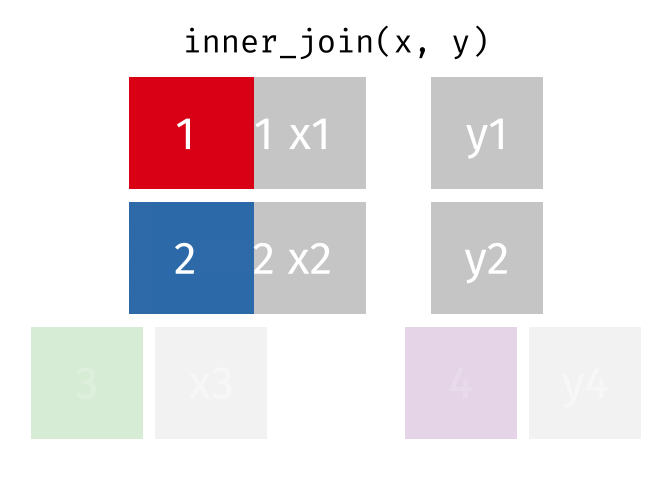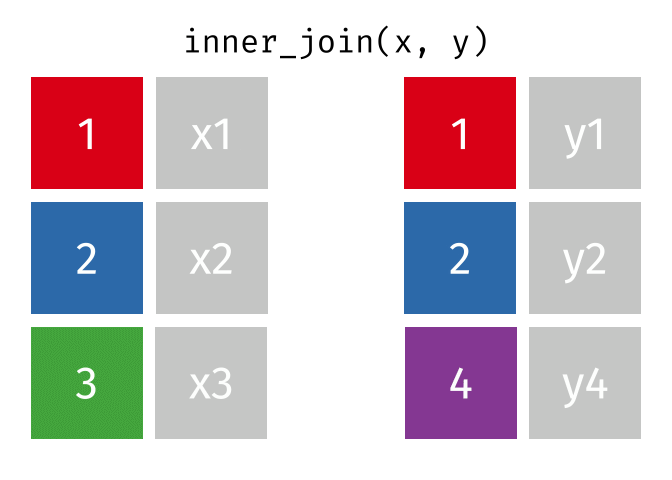
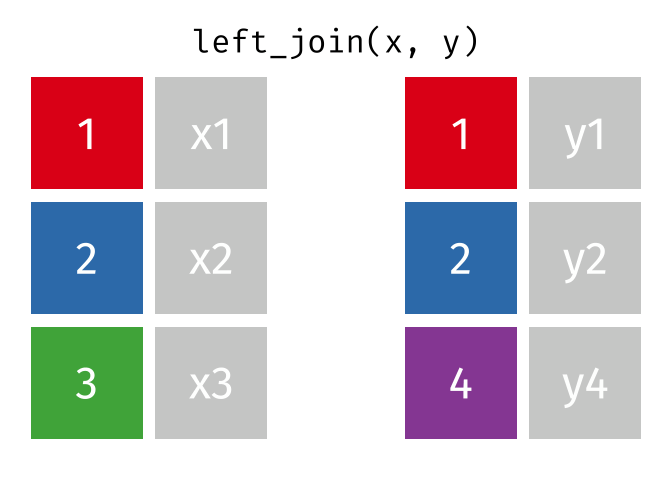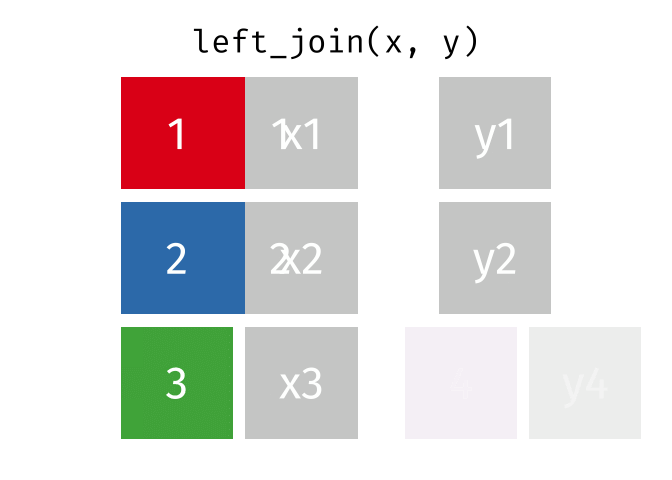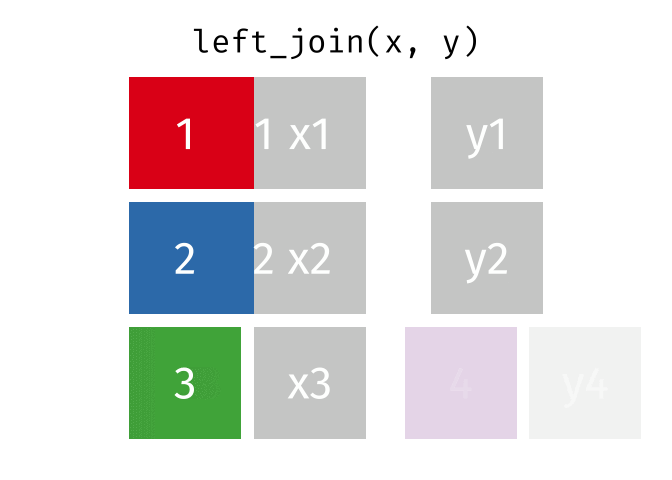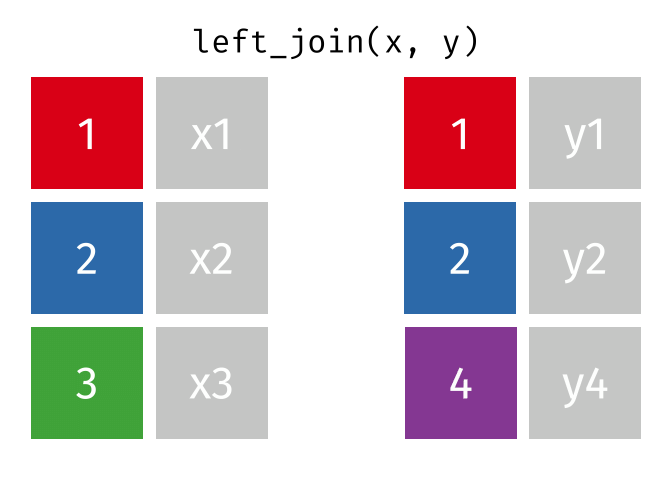
- Joining and intersecting tibbles
- Selection and manipulation
- In a workflow typically follows reshaping operations from
tidyr - Fast, by writing key pieces in C++ (using
Rcpp) - Standard interfaces to database (
dbplyr) ordata.table(dtplyr).
Preparing data is the most time consuming part of of data analysis!
Essential part of understanding data, hard to avoid.